
UKCA Mark & Alternative routes to market – a risk or an important step towards global regulation?
Next under the spotlight of this series is alternative routes to market – what are the proposals, pitfalls, and opportunities?
Chapter 14 of the public consultation covered two key areas:
Section 72 – MDSAP and domestic assurance
Section 73 – Pathway for Innovative MedTech
Medical Device Single Audit Program (MDSAP)
MHRA proposed to proceed with requiring UK Approved Bodies to ‘consider’ MDSAP assessments, however it is not a mandatory route for manufacturers to take.
We know that MHRA are official observers of the MDSAP. It is expected that they will apply to full membership in the near future, further cementing the global approach they are keen to support.
This would allow for a reduction in the review needed on the QMS aspects of a technical file – the ideas being for shortened review to be conducted by the Approved Body. The global use of resources is definitely an appealing idea…
Domestic Assurance
The domestic assurance proposal goes a step further – this looks to offer abridged reviews for products with regulatory approvals from certain jurisdictions. Whilst those jurisdictions have not been identified in the response, an educated guess would be that FDA, Health Canada, EU and TGA approvals will be on that list!
This proposal by the MHRA offers one of the key opportunities for a ‘Global Britain’, allowing for these alternative routes allows for market flexibility, increased patient access to devices, global synergies and economic benefits for the UK. If done well and consistently this could be the beginning of a truly global conformity route. This is an exciting proposition and one that will hopefully come to fruition.
However, there are some challenges and questions to be asked.
- Is there a risk to patient safety? How do we ensure enough ‘domestic assurance’ is done?
- How can this be done consistently? Approved bodies are the ones that have accept the risk, how far will they abridge their assessments?
- Is the capacity there to be able to achieve this?
- What will the data gaps be on accepting other approvals? Where do the current frameworks differ? Are these a critical mismatch that will prevent this approach from succeeding?
- Will there be a push for mutual recognitions and, therefore similar acceptance of the UKCA mark in the future?
What could the routes to UKCA mark look like?
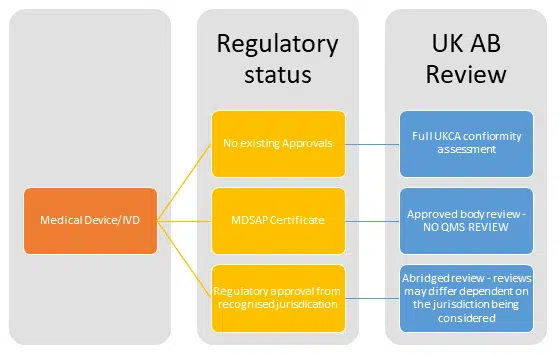
Do these new approaches work with rest of the system?
In the response we see a lot of alignment/synergy with the EU MDR & IVDR – increased scrutiny across the lifecycle, increased Approved Body activity, and increased Regulator activity.
So how do these abridged reviews fit in?
Post-market – how does this fit in? Will these routes to market then feed into the system as proposed, or will there be additional requirements?
Is there a huge opportunity waiting to be realised? Extensive engagement is needed with the Approved Bodies to truly understand the implications and the process challenges.
Should domestic assurance sit outside of the Approved body system, do we need abridged reviews, or should it be outright acceptance of trusted global conformity assessments? This would reduce burden, but potentially opens up the UK healthcare system and patients to greater risk. How would the post-market requirements be achieved? Who would be responsible for PSURs for example – who would do the review? Would this be good or bad for the UK market?
A lot of questions, a lot of opportunities to be a gateway. However, is the current proposal too risk adverse?
Innovative MedTech & IVDs
The response sets out that MHRA intended to put in place a route where MHRA can grant initial market approval.
The route would be ringfenced to specific and clearly defined circumstances. Work is already ongoing to consider an end–to–end process that would be established in partnership with the National Institute for Health and Care Excellence.
The UK has a lot of options for innovators, not all of it is clear or accessible – If MHRA and NICE can deliver a pathway that truly supports innovators through the process, allowing for safety and timely access to patients, then this will be a significant ‘win’ for the system. It also offers a unique opportunity for those products to generate Real World Data at an earlier stage – earlier patient feedback, Healthcare professional feedback and analysis of efficiency and cost impact for the system – all vital data.
Whilst there are lots of benefits with this type of pathway the fundamental point is – Patient safety must not suffer, and risk benefit assessment must be the underpinning principle.
Globally, the Breakthrough Device Program shows there is a need and market for such a process. This would further position the MHRA as a key regulatory leader, and the UK as an attractive innovation hub.
Again, the detail is not there. We need to know where the pathway leads, will it be an MHRA conformity assessment, or will the products feedback into the Approved Body system? This will have implications for application times and review capacity.
More detail is needed around selection and how companies navigate this pathway – what are the cost implications? Does it offer huge improvements in ‘time to market’, how will these products be reviewed?
Are the partners in the UK system ready to hand over some responsibility to others?
Will this lead to the time efficiencies the system so badly needs?
Can Global regulation deliver for MedTech & IVDs?
What is the systems ‘risk appetite’?
Next steps
The response to the consultation is just that – this is not official policy yet and the draft regulations are in progress. We will have to await further details before we can fully review and discuss the impact on the sector.
None of these processes will be quick to implement – understanding the projected timeline for these changes is critical.
We hope this has been an interesting read. If you have any more questions about the MHRA response and the future for the UK please do get in touch. Also keep an eye out for our article on MDSAP coming soon.
