A 510(k) is the technical dossier required by the US Food and Drug Administration (FDA) to sell a medium-risk medical device or IVD in the United States.
The 510k and PMA submission routes depend on the Device Class
Devices are subject to a comprehensive set of regulatory authorities called general controls that are applicable to all classes of devices
Submission type or Exemption: 510k or 510k Exempt
Class II- moderate risk
Higher risk than class I
Devices for which general controls, by themselves, are insufficient to provide reasonable assurance of the safety and effectiveness of the device, and for which there is sufficient information to establish special controls to provide such assurance
submission type or Exemption: 510(k) or 510(k) Exempt
Class III- hightest risk
Sustain or support life, are implanted, or present potential unreasonable risk of illness or injury
Devices for which general controls, by themselves, are insufficient and for which there is insufficient information to establish special controls to provide reasonable assurance of the safety and effectiveness of the device. Class III devices typically require premarket approval
Submission type ot exemption: PMA
What is the 510k route, also called the Pre-Market Notification?
If you wish to place a device on the US market and a Premarket Approval Application is not required – you must submit a 510(k) to the FDA *note there are exemptions & limitations to this ‘.9 of the device classification regulation chapters (21 CFR 862.9, 21 CFR 864.9)’
The main principle of this submission is to demonstrate that your product is as ‘safe and effective’ (substantially equivalent) to a device that is legally marketed in the US. The legally marketed device or devices are referred to as the predicate devices(s).
You cannot market your device until the FDA have deemed it to be substantially equivalent – this decision is usually made within 90 days and is solely based on the information you provide in your submission.
510k submission cleared by the FDA are published – usually in the first week of the following month.
Class I- lowest risk
Devices are subject to a comprehensive set of regulatory authorities called general controls that are applicable to all classes of devices
Class II- moderate risk
Devices for which controls, by themselves, are insufficient and for which there is insufficient information to establish special controls to provide reasonable assurance of the safety and effectiveness of the device. Class III devices typically require premarket approval.
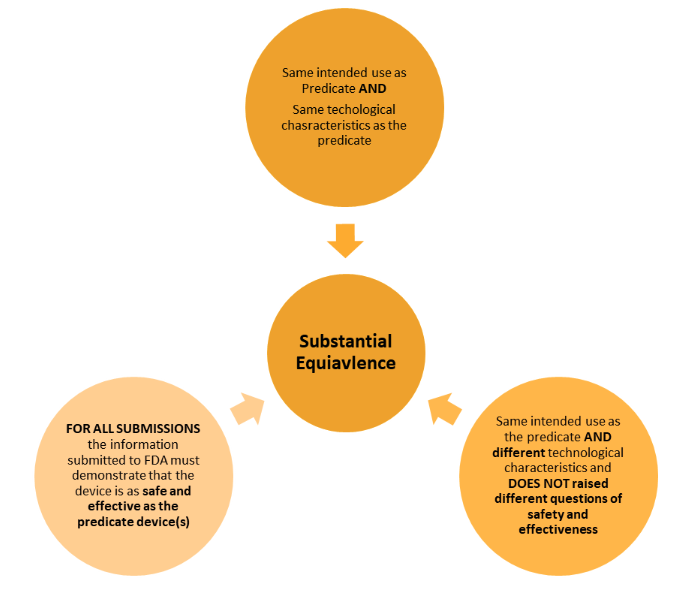
What are the criteria for substantial equivalence in US with the 510k submission ?
What counts as a predicate device?
A legally marketed device that was marketed prior to May 28th 1976
A device that had been reclassified from Class III to class II or I
A device that was found to be substantially equivalent through a 510(k) submission
A device granted marketing authorisation through the De Novo classification process 510k
It is possible to use more than one predicate, times this is utilized include:
When combining features from two or more predicate devices with the same intended use into a single new device
Where the device under evaluation has more than one intended use
Where a device has more than one indication for use under the same intended use
If there is no appropriate predicate device, then manufacturers will need to submit a De Novo request or for high risk products a Premarket Approval may be required.
The approach to demonstrating equivalence is different to the EU/UKand has two key considerations: intended use and technological characteristics.
Below shows the two different ways in demonstrating this.
There are less strict requirements within the regulations when compared to the EU approach – however the principles are very much the same. The similarities and the differences must be clearly set out and evaluated.
There must be clear evidence to demonstrate the use of the predicate device(s) is valid and that there is assurance that the safety and effectiveness are substantially equivalent.
The onus is on the manufacturer to ensure this is presented in a robust and scientifically valid way.
This installment will turn the attention to the US and the FDA’s use of equivalence in us
Common reasons for Not Substantially Equivalent (NSE) decision
These fall in to 2 categories – (1) FDA decision that the device is Class III and cannot be reviewed through 510(k); (2) inadequacies in evidence provided in the submission
Lack of predicate
New intended use
Different technological characteristics with questions over saftey of effectiveness
insuficient evidence provided
Lack of performance data
If an NSE decision is given – the manufacturer is given the chance to respond to the FDA determination & the deficiencies noted.
Take home message:
This is a useful route for the US market that offers more timely access to market
It should not be seen as a short cut – assurance around safety and performance is critical. Provide clear and robust evidence in the submission. Only use legally marketed devices as a predicate
We hope this has helped set out the requirements and considerations when looking to use the FDA’s 510(k) route. For further guidance on a evaluating substantial equivalence for your device you can refer to the FDA’s guidance
How ECLEVAR Can help you with the 510k submission routes:
Clinical project manager
Add Your Tooltip Text Here
Data protection officer
Add Your Tooltip Text Here
Data manager
Add Your Tooltip Text Here
Data engineers
Add Your Tooltip Text Here
Senior Statistician
Add Your Tooltip Text Here
Clinical Nurses
Add Your Tooltip Text Here
Medical advisors
Add Your Tooltip Text Here
Reforming Clinical Evaluation of Medical Devices in Europe