Eine 510(k)-Zulassung ist das technische Dossier, das die US Food and Drug Administration (FDA) für den Verkauf eines Medizinprodukts oder IVD mit mittlerem Risiko in den Vereinigten Staaten verlangt.
Wenn Sie ein Produkt in den USA auf den Markt bringen möchten und kein Antrag auf Vorabgenehmigung erforderlich ist, müssen Sie bei der FDA einen 510(k)-Antrag einreichen *beachten Sie, dass es hierzu Ausnahmen und Beschränkungen gibt ‚.9 der Kapitel über die Klassifizierung von Produkten (21 CFR 862.9, 21 CFR 864.9)‘
Das Hauptprinzip dieser Einreichung besteht darin, nachzuweisen, dass Ihr Produkt so sicher und wirksam“ (im Wesentlichen gleichwertig) ist wie ein Produkt, das in den USA legal vermarktet wird. Das rechtmäßig in Verkehr gebrachte Produkt oder die rechtmäßig in Verkehr gebrachten Produkte werden als Prädikat(e) bezeichnet.
Sie können Ihr Produkt erst vermarkten, wenn die FDA es als im Wesentlichen gleichwertig eingestuft hat. Diese Entscheidung wird in der Regel innerhalb von 90 Tagen getroffen und basiert ausschließlich auf den Informationen, die Sie in Ihrem Antrag angegeben haben.
Von der FDA genehmigte 510k-Anträge werden veröffentlicht – in der Regel in der ersten Woche des Folgemonats.
Was gilt als Prädikatsprodukt?
Es ist möglich, mehr als ein Prädikat zu verwenden, wovon unter anderem Gebrauch gemacht wird:
Im Folgenden werden die beiden verschiedenen Arten des Nachweises dargestellt.
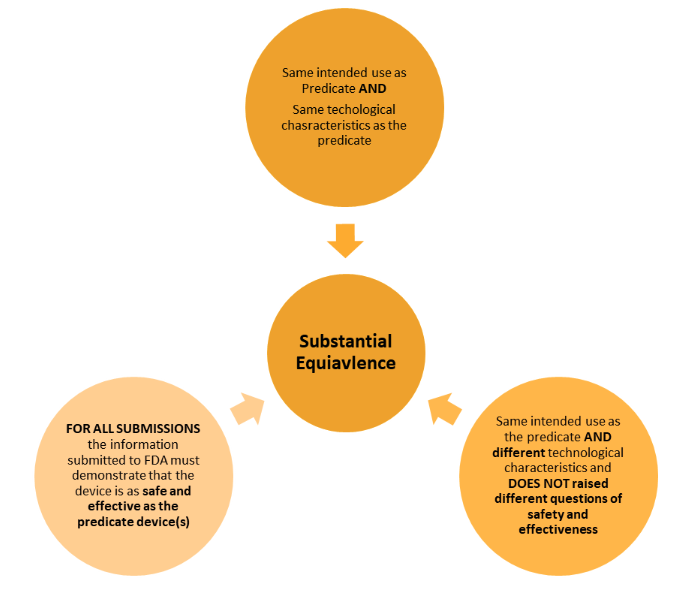
Im Vergleich zum EU-Ansatz sind die Anforderungen in den Verordnungen weniger streng – die Grundsätze sind jedoch weitgehend die gleichen. Die Ähnlichkeiten und Unterschiede müssen klar dargelegt und bewertet werden.
Es muss eindeutig nachgewiesen werden, dass die Verwendung des/der Prädikatprodukts/Präparate zulässig ist und dass Sicherheit und Wirksamkeit im Wesentlichen gleichwertig sind.
Es liegt in der Verantwortung des Herstellers, dies auf solide und wissenschaftlich fundierte Weise darzulegen.
In dieser Folge geht es um die USA und die Verwendung des Begriffs der Gleichwertigkeit durch die FDA in den USA.
Häufige Gründe für die Entscheidung „Nicht substanziell gleichwertig“ (Not Substantially Equivalent, NSE)
Diese lassen sich in zwei Kategorien einteilen – (1) Entscheidung der FDA, dass das Produkt der Klasse III angehört und nicht nach 510(k) geprüft werden kann; (2) unzureichende Nachweise in der Einreichung
Wird eine NSE-Entscheidung getroffen, erhält der Hersteller die Möglichkeit, auf die FDA-Entscheidung und die festgestellten Mängel zu reagieren.
Die Botschaft zum Mitnehmen:
Dies ist ein nützlicher Weg für den US-Markt, der eine schnellere Markteinführung ermöglicht DeepL
Er sollte nicht als Abkürzung gesehen werden – Sicherheit und Leistung sind entscheidend.
Legen Sie in Ihrem Antrag klare und solide Nachweise vor. Verwenden Sie nur legal vermarktete Produkte als Prädikat.
Wir hoffen, dass diese Ausführungen dazu beigetragen haben, die Anforderungen und Überlegungen bei der Nutzung der 510(k)-Route der FDA darzulegen.
Weitere Hinweise zur Bewertung der substanziellen Äquivalenz für Ihr Produkt finden Sie in den Leitlinien der FDA

Abonnieren Sie unseren Newsletter
30 Minuten kostenlose Beratung
Während 30 Minuten beantworten unsere Eclevar-Experten Ihre Fragen und führen Sie durch die nächsten Schritte!
BESUCHE UNS
ECLEVAR GMBH
ERFURT, Erfurt Hauptbahnhof
4th, 5th floor
Bahnhofstr. 38 Erfurt
99084
BLEIBEN SIE AUF DEM LAUFENDEN