Serie von Zusammenfassungen der MDCG-Leitfaden-Dokumente:
MDCG 2019-9 Rev.1 – Zusammenfassung der Sicherheit und klinischen Leistung Ein Leitfaden für Hersteller und benannte Stellen
Was ist SSCP, wer ist involviert, und was ist die Intention?
Mit der MDR 2017/745 Art. 32 müssen Hersteller für implantierbare Produkte und für Produkte der Klasse III, die keine Sonderanfertigungen oder Prüfprodukte sind, einen Kurzbericht über Sicherheit und klinische Leistung (SSCP) vorlegen.
Dabei handelt es sich um eine Zusammenfassung der klinischen Daten und anderer Informationen über die Sicherheit und klinische Leistung des Medizinprodukts.
Die SSCP ist ein zusätzliches Dokument zur Gebrauchsanweisung (IFU), zu den Implantatpass oder zu anderen obligatorischen Dokumenten. Das SSCP hat objektiv zu sein und fasst günstige und ungünstige Daten zusammen.
Die SSCP richtet sich an professionelle Anwender, aber auch an Patienten, wenn dies erforderlich ist (z. B. wenn ein Implantatpass erforderlich ist, wenn der Patient der Anwender des Produkts ist), und zwar in den Landessprachen der Mitgliedstaaten, in denen das Produkt in Verkehr gebracht wird, und mit einer für Laien verständlichen Formulierung.
Die benannte Stelle (NB) ist verpflichtet, das SSCP zu validieren und in EUDAMED hochzuladen, so dass es öffentlich und kostenlos im PDF-Format zur Verfügung steht.
Dies ist ein Schritt, der mit der MDR 2017/745 eingeführt wurde, um die Transparenz und die Information der Öffentlichkeit zu verbessern.
Die MDCG hat diesen Leitfaden erstellt, um die Mindestanforderungen an Darstellung, Inhalt und Validierung der SSCP zu unterstützen und zu klären.
Die ursprüngliche Version wurde im September 2019 bereitgestellt, wurde aber kürzlich aktualisiert, um einige Klarstellungen zur Basis-UDI-DI in Verbindung mit dem SSCP in EUDAMED (#3.1 Seiten 12, 13) zu geben, und die in der Vorlage zu erfassende Referenznummer des Herstellers wurde hinzugefügt.
Allgemeine Anforderungen und Empfehlungen für die SSCP
Für die Aktualität des SSCP ist der Hersteller verantwortlich.
Quellen für den SSCP sind:
- Technische Dokumentation (TD) wie Berichte über die Prüfung und Validierung des Produktdesigns, der Risikomanagementbericht/die Risikomanagementakte, der Bericht über die klinische Bewertung sowie Pläne und Berichte über die Überwachung nach dem Inverkehrbringen (PMS) und die klinische Nachbeobachtung nach dem Inverkehrbringen (PMCF), PSUR.
- Da die IFU auch Informationen aus diesen Quellen enthält, kann sie gegebenenfalls als Quelle für die SSCP verwendet werden.
Eine eindeutige SSCP-Referenznummer:
- wird vom Hersteller im Rahmen des Managementsystems des Herstellers vergeben
- bleibt während der gesamten Lebensdauer der SSCP gleich
- dient in Verbindung mit der SRN des Herstellers als eindeutige Identifizierung der SSCP in EUDAMED und in der EU
- die SSCP muss regelmäßig aktualisiert werden und in EUDAMED auf dem neuesten Stand sein.
Die Benannte Stelle (NB) ist die einzige Partei, die die SSCP in EUDAMED hochladen kann
Ist mehr als ein NB beteiligt, ist der für die Validierung der technischen Unterlagen zuständige NB für die Validierung der SSCP zuständig.
Neu ist auch, dass die IFU alle Informationen enthalten muss, die zum Auffinden der SSCP in EUDAMED erforderlich sind. Die IFU muss Folgendes enthalten (Seite 6):
- „Es ist anzugeben, dass die SSCP in der europäischen Datenbank für Medizinprodukte (Eudamed) verfügbar ist, in der sie mit der Basis-UDI-DI verknüpft ist.
- Sie sollte die URL der öffentlichen Eudamed-Website angeben: https://ec.europa.eu/tools/eudamed
- Es sollte der Code der Basis-UDI-DI angegeben werden. Alternativ kann auch ein anderes Meta-Datenformat angegeben werden, sofern es für die eindeutige Suche und das Auffinden der vorgesehenen SSCP in Eudamed verwendet werden kann.“
Je nachdem, ob es sich bei dem Nutzer um Angehörige der Gesundheitsberufe oder um Patienten handelt, können unterschiedliche Sprachanforderungen an eine IFU gestellt werden, und die Mitgliedstaaten haben möglicherweise unterschiedliche Anforderungen. Dies gilt auch für die SSCP. Dies gilt auch für die SSCP.
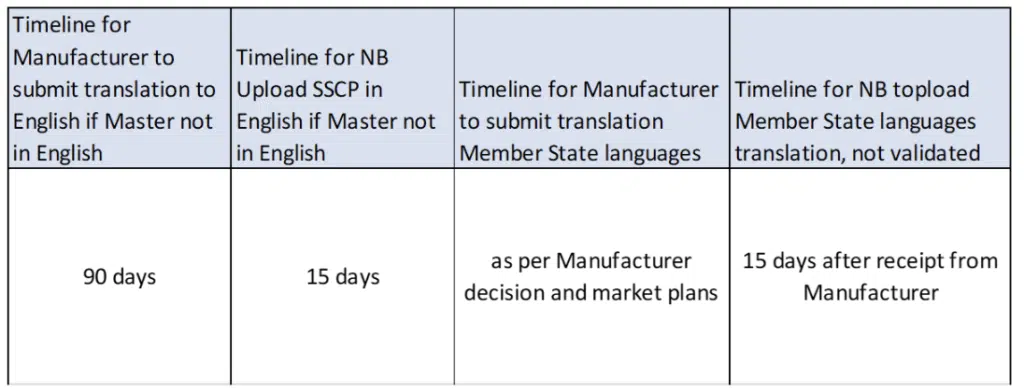
Die SSCP kann in einer vom NB akzeptierten Sprache der EU-Mitgliedstaaten abgefasst sein (Master-SSCP), aber wenn dies eine andere Sprache als Englisch ist, muss der Hersteller dem NB zusätzlich eine übersetzte englische Master-Version vorlegen. Die Master-SSCP wird vom NB validiert und dann in EUDAMED hochgeladen.
Der Hersteller ist für die Übersetzung ins Englische sowie für die Übersetzung in andere EU-Sprachen, in denen das Produkt in Verkehr gebracht werden soll, verantwortlich, einschließlich der Überprüfung der Korrektheit der Übersetzung. Der NB wird die Übersetzungen in EUDAMED hochladen, aber nicht validieren.
Für jede Sprache sollte eine separate SSCP erstellt werden, einschließlich einer Erklärung, in welcher Sprache die SSCP vom NB validiert wurde.
SSCP – Anforderungen, Validierung, Upload, Übersetzungen und Zuständigkeiten
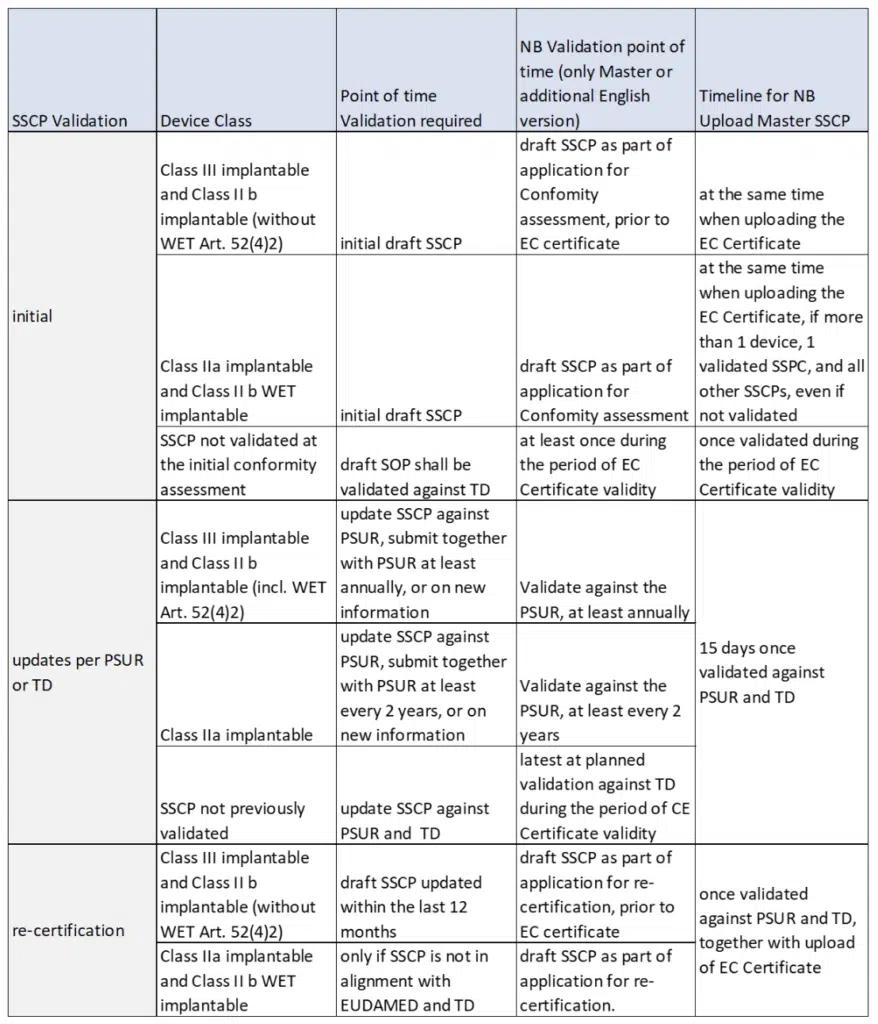
Der SSCP-Entwurf wird der benannten Stelle (NB) zur Validierung vorgelegt:
- bei Erstvalidierung für die Konformitätsbewertung oder für die erste SSCP für alte oder veraltete Geräte
- bei Re-Zertifizierung der Konformitätsbewertung, oder wenn keine validierte Version des SSCP verfügbar ist
- wenn der PMCF-Bewertungsbericht und der regelmäßige aktualisierte Sicherheitsbericht (PSUR) aktualisiert werden
- wenn neue Informationen verfügbar sind
Im Folgenden finden Sie eine Zusammenfassung der Anforderungen und Aufgaben für Benannte Stellen und Hersteller
Management von Übersetzungen:
Anleitungen für jeden der erforderlichen Abschnitte des SSCP-Dokuments
Die Einzelheiten für eine SSCP-Vorlage sind im Anhang aufgeführt: Vorlage für die SSCP, die die erforderlichen Mindestinformationen enthält.
Die MDCG hat für jeden erforderlichen Abschnitt eine Anleitung zur Verfügung gestellt. Diese sind sehr informativ und geben Antworten und Erläuterungen in Bezug auf die erwarteten Informationen in den Abschnitten der SSCP:
- Kennzeichnung des Produkts und des Herstellers, einschließlich der Basis-UDI-DI und, falls bereits vergeben, der SRN
- Die Zweckbestimmung des Produkts sowie alle Indikationen, Kontraindikationen und Zielpopulationen
- eine Beschreibung des Produkts, einschließlich eines Verweises auf frühere Generationen oder Varianten, sofern solche existieren, und einer Beschreibung der Unterschiede sowie gegebenenfalls eine Beschreibung von Zubehör, anderen Produkten und Produkten, die in Kombination mit dem Produkt verwendet werden sollen
- Informationen über etwaige Restrisiken und unerwünschte Wirkungen, Warnhinweise und Vorsichtsmaßnahmen
- Zusammenfassung der klinischen Bewertung gemäß Anhang XIV und relevante Informationen über die klinische Nachbeobachtung nach dem Inverkehrbringen
- Mögliche diagnostische oder therapeutische Alternativen
- Empfohlenes Profil und Schulung für die Anwender
- Verweis auf alle angewandten harmonisierten Normen und gemeinsamen Spezifikationen (CS)
- Historie der Überarbeitung
Die MDCG empfiehlt, die Informationen für die vorgesehenen Nutzer/Angehörige der Gesundheitsberufe und für die Patienten in zwei Teilen der SSCP zu trennen.
Andere Hilfreiche MDCG Leitlinien
- MDCG2019-3 Rev. 1 Ausnahmen vom Konsultationsverfahren zur klinischen Bewertung Auslegung von Artikel 54 Absatz 2 Buchstabe b
- MDCG 2019-8 v2 Leitfaden Implantatpass zur Anwendung von Artikel 18 der Verordnung (EU) 2017/745 über Medizinprodukte
- MDCG 2019-11 Qualification and classification of software – Regulation (EU) 2017/745 and Regulation (EU) 2017/746
- MDCG 2019-16 rev.1 Leitlinie zur Cybersicherheit für Medizinprodukte
- MDCG 2020-1 Leitlinie zur klinischen Bewertung (MDR) / Leistungsbewertung (IVDR) von Software für Medizinprodukte
- MDCG 2020-6 Leitlinie zum ausreichenden klinischen Nachweis für Altprodukte
- MDCG 2020-7 Leitlinie zur Vorlage für den PMCF-Plan
- MDCG 2020-8 Leitfaden zur Vorlage für den PMCF-Bewertungsbericht
- MDCG 2020-10/1 Leitfaden zur Sicherheitsberichterstattung bei klinischen Prüfungen und MDCG 2020-10/2 Formular für den zusammenfassenden Sicherheitsbericht über klinische Prüfungen v1.0
- MDCG 2020-13 Vorlage für den Bericht zur Bewertung der klinischen Bewertung (und MEDDEV 2.7/1 rev. 4)
- Die MDCG 2021-1 Rev.1 Leitlinien für harmonisierte Verwaltungspraktiken und alternative technische Lösungen, bis EUDAMED voll funktionsfähig ist (bzw. Verweise auf: Art. 70, Art. 73, Art. 74, Art. 75, Art. 76, Art. 77, Art. 78, Art. 80), siehe auch unseren Blog: Zusammenfassung des MDCG-Leitfadens MDCG 2021-20
- MDCG 2021-6 Verordnung (EU) 2017/745 – Fragen und Antworten zur klinischen Prüfung
- MDCG 2021-8 Dokumente für die Beantragung oder Notifikation einer Klinische Prüfung, siehe auch unseren Blog: Zusammenfassung des MDCG-Leitfadens MDCG 2021-8
- MDCG 2021-11 Leitfaden zum Implantatpass- Produkttypen
- MDCG 2021-20 Anleitung zur Erstellung der CIV-ID für klinische Prüfungen unter der MDR 2017/745, siehe auch unseren Blog: Zusammenfassung des MDCG-Leitfadens MDCG 2021-20
- MDCG 2021-25 Anwendung der MDR-Anforderungen auf “ Legacy Produkte “ und auf Produkte, die vor dem 26. Mai 2021 in Übereinstimmung mit den Richtlinien 90/385/EWG oder 93/42/EWG in Verkehr gebracht wurden
- MDCG 2021-28 Wesentliche Änderung einer klinischen Prüfung unter der Medizinprodukteverordnung, siehe auch unseren Blog: Zusammenfassung des MDCG-Leitfadens MDCG 2021-28
- MDCG-Leitlinien im Abschnitt Benannte Stellen
- MDCG-Leitlinien im Abschnitt Unique Device Identifier (UDI)
Es werden hilfreiche Leitlinien für die Klassifizierung von Medizinprodukten und Kombinationsprodukten zur Verfügung gestellt:
- MDCG 2022-5 Leitfaden zur Abgrenzung zwischen Medizinprodukten und Arzneimitteln gemäß Verordnung (EU) 2017/745 über Medizinprodukte
- MDCG 2021-24 Leitfaden zur Klassifizierung von Medizinprodukten
- Informationsaustausch zwischen den für Medizinprodukte zuständigen Behörden über Grenzfälle und Klassifizierungen Helsinki-Verfahren 2021
Zusätzlich werden hilfreiche Leitlinien für die Registrierung von „Akteuren“ im Rahmen der MDR bereitgestellt:
- MDCG 2020-15: MDCG-Positionspapier über die Verwendung des EUDAMED-Moduls zur Registrierung von Akteuren und der einheitlichen Registrierungsnummer (SRN) in den Mitgliedstaaten
- MDCG 2021-13 rev.1: Fragen und Antworten zu den Verpflichtungen und damit verbundenen Regeln für die Registrierung in EUDAMED von anderen Akteuren als Herstellern, Bevollmächtigten und Importeuren, die den Verpflichtungen von Artikel 31 MDR und Artikel 28 IVDR unterliegen
- MDCG 2022-12 Leitfaden für harmonisierte Verwaltungspraktiken und alternative technische Lösungen, bis Eudamed voll funktionsfähig ist (für die Verordnung (EU) 2017/746 über In-vitro-Diagnostika)
- MDCG 2019-5 Registrierung von Altgeräten in EUDAMED
- MDCG 2019-4 Zeitpläne für die Registrierung von Produktdatenelementen in EUDAMED
Leitfaden zur Produktnomenklatur:
- MDCG 2018-2 Künftige EU-Nomenklatur für Medizinprodukte – Beschreibung der Anforderungen
- MDCG 2021-12 FAQ zur Europäischen Nomenklatur für Medizinprodukte (EMDN)
- Das EMDN – Die Nomenklatur zur Verwendung in EUDAMED
- Die CND-Nomenklatur – Hintergrund und allgemeine Grundsätze
Ich danke Ihnen für Ihre Zeit. Wenn Sie dies interessant fanden, lesen Sie bitte unseren nächsten Blog über die Zusammenfassungen der MDCG-Dokumente: MDCG 2022-5 Leitfaden zur Abgrenzung zwischen Medizinprodukten und Arzneimitteln gemäß Verordnung (EU) 2017/745 über Medizinprodukte