Un 510(k) est le dossier technique requis par la Food and Drug Administration (FDA) américaine pour vendre un dispositif médical ou un DIV à risque moyen aux États-Unis.
Si vous souhaitez mettre un dispositif sur le marché américain et qu’une demande d’autorisation de mise sur le marché n’est pas nécessaire, vous devez soumettre un 510(k) à la FDA. *Notez qu’il existe des exemptions et des limitations à cette demande ‘.9 des chapitres de la réglementation sur la classification des dispositifs (21 CFR 862.9, 21 CFR 864.9)’.
Le principe principal de cette soumission est de démontrer que votre produit est aussi « sûr et efficace » (substantiellement équivalent) qu’un dispositif légalement commercialisé aux États-Unis. Le(s) dispositif(s) légalement commercialisé(s) est (sont) appelé(s) dispositif(s) prédicat(s).
Vous ne pouvez pas commercialiser votre dispositif tant que la FDA ne l’a pas jugé substantiellement équivalent – cette décision est généralement prise dans les 90 jours et repose uniquement sur les informations que vous avez fournies dans votre demande.
Les demandes 510k approuvées par la FDA sont publiées – généralement au cours de la première semaine du mois suivant.
Qu’est-ce qu’un dispositif prédicat ?
Il est possible d’utiliser plus d’un prédicat, notamment dans les cas suivants :
S’il n’y a pas de dispositif prédicat approprié, les fabricants devront soumettre une demande De Novo ou, pour les produits à haut risque, une approbation avant commercialisation peut être requise.
L’approche pour démontrer l’équivalence est différente de celle de l’UE/du Royaume-Uni et comporte deux considérations clés : l’utilisation prévue et les caractéristiques technologiques.
Le tableau ci-dessous montre les deux différentes manières de démontrer l’équivalence.
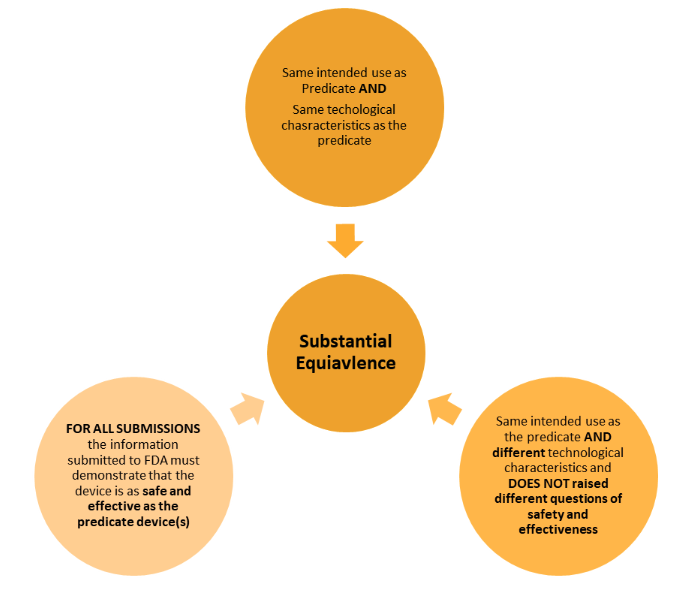
Les exigences de la réglementation sont moins strictes que celles de l’approche européenne, mais les principes sont très similaires. Les similitudes et les différences doivent être clairement exposées et évaluées.
Il doit y avoir des preuves claires pour démontrer que l’utilisation du ou des dispositifs prédicats est valide et qu’il existe une assurance que la sécurité et l’efficacité sont substantiellement équivalentes.
Il incombe au fabricant de s’assurer que ces éléments sont présentés d’une manière solide et scientifiquement valide.
Dans cet article, nous allons nous intéresser aux États-Unis et à l’utilisation de l’équivalence par la FDA.
Raisons courantes d’une décision de non-équivalence substantielle (NSE)
Elles se répartissent en deux catégories : (1) décision de la FDA selon laquelle le dispositif est de classe III et ne peut pas être examiné par le biais de la procédure 510(k) ; (2) insuffisance des preuves fournies dans la soumission.
Si une décision NSE est rendue, le fabricant a la possibilité de répondre à la détermination de la FDA et aux déficiences constatées.
Message à retenir :
Il s’agit d’une voie utile pour le marché américain qui permet une mise sur le marché plus rapide (DeepL).
Elle ne doit pas être considérée comme un raccourci – l’assurance de la sécurité et des performances est essentielle.
Fournir des preuves claires et solides dans la soumission. N’utilisez que des dispositifs légalement commercialisés comme prédicat.
Nous espérons que cela vous a aidé à définir les exigences et les considérations à prendre en compte lorsque vous cherchez à utiliser la procédure 510(k) de la FDA.
Pour plus d’informations sur l’évaluation de l’équivalence en substance de votre dispositif, vous pouvez vous référer au guide de la FDA.

