510(k)とは、米国において中等度のリスクに
分類される医療機器およびIVD(体外診断機器)を販売するうえで、
米国食品医薬局(FDA)から認可を得るための申請ルートです。
害を及ぼす可能性が最小のもの
すべての医療機器に適用されるGeneral Controls(一般管理)と呼ばれる包括的な規制の対象となる医療機器。
申請の種類または免除:510kまたは510k免除
クラスIよりリスクが高いもの
General Controlsの規定だけでは医療機器の安全性と有効性の妥当な担保ができない医療機器であり、そのために、Special Controls(特別管理)項目を設定して安全性と有効性を保証する必要のある医療機器。
生命を維持するための機器、インプラント製品、および重大な傷病を起こす可能性のあるリスクがある製品。
General Controlsの規定だけでは医療機器の安全性と有効性の妥当な担保ができない医療機器であり、そのために、Special Controls(特別管理)項目を設定して安全性と有効性を保証する必要のある医療機器。通常の場合、クラスIII製品はPMA(市販前承認)審査が必要となる。
申請の種類または免除:PMA
米国で医療機器を販売したいと考えており、PMA(市販前承認)申請が必要でない場合は、
FDAに510(k)申請を提出しなければならなりません。*注:510(k)には免除と制限があります(21 CFR 862.9, 21 CFR 864.9参照)。
510k申請の主な原則は、すでに米国内にて合法的に販売されている医療機器と「安全性と有効性」において実質的に同等であることを示すことです。この合法的に既に販売されている先行品のことをPredicate Deviceと呼びます。
貴社の製品に関して、FDAがこのPredicate Deviceと実質的に同等の製品であると判断を下すまでは販売することができません。この決定は、通常、90日以内に行われ、貴社が提出した資料のみに基づいて判断されます。
FDAで認可された510kの申請は公表されます。通常、認可の翌月の第1週に公表されます。
どんなものがPredicate Deviceとなりますか?
1976年5月28日以前に合法的に販売された機器
クラスIIIからクラスIIまたはIに分類し直された機器
510(k)申請により実質的同等性が示された機器
De Novo(デノボ)分類の申請ルートで510k販売認可を受けた機器
複数のPredicate Deviceを使用することも可能です。複数のPredicate Deviceを使用する場合としては次のような場合です:
同じ使用目的の二つ以上のPredicate Deviceの機能を一つの機器に統合した製品の場合
審査対象の医療機器が複数の使用目的を有する場合
機器が同じ使用目的の下で、複数の適応症がある場合
該当するPredicate Deviceがない場合、製造者はDe Novoによる申請を行う必要があります。また、高リスク製の場合は、PMAの申請が必要になる場合もあります。
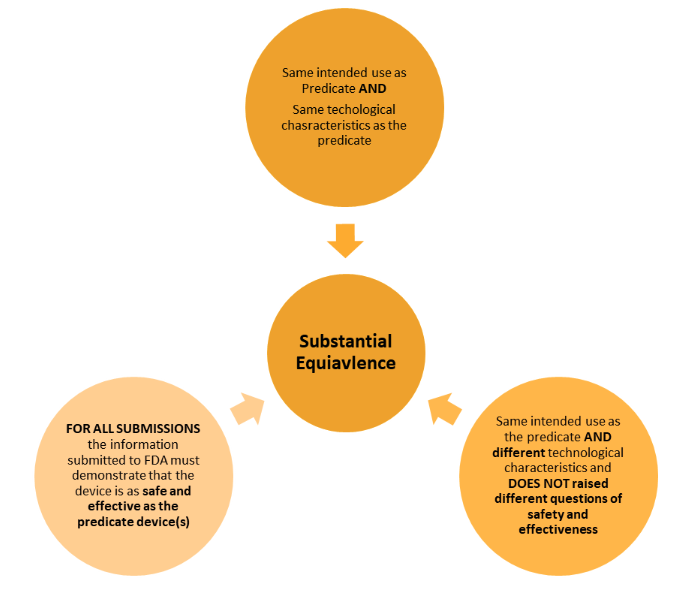
同等性を証明する方法に関しては、EUや英国の場合とは異なり、使用目的と技術的特徴の2つの点について考慮する必要があります。
以下は、同等性を示す二つの方法です。
実質的同等性
Predicate Deviceと同じ使用目的ならびに同じ技術的特性
Predicate Deviceと同じで使用目的で、技術的特性は異なるが、安全性と有効性に関して疑義が生じない
“すべての申請において、
FDAに提出した情報は申請した機器がPredicate Deviceと同等の安全性と有効性があることを示すものでなければなりません。”
EUの規則と比べて、FDAの規則の要求項目はそれほど厳しいものではありませんが、原則はほとんど同じです。類似点と相違点を明確に示し、評価しなければなりません。
選択したPredicate Deviceを同等製品として比較することの妥当性を明確に示すとともに、安全性と有効性が実質的に同等であることを保証する明確な証拠を示す必要があります。
このような根拠をしっかりとして科学的に有効な方法で提示する責任は製造者側にあります。
ここで記載している情報は、米国における同等性についてのFDAと米国の考え方に着目するものです。
「実質的な同等性とは言えない」(NSE)と判断される一般的な理由
この一般的な理由は次の二つのカテゴリーに分かれます:
(1)医療機器がクラスIIIに該当し、510(k)としての審査ができないと判断する場合;
(2)申請書類のエビデンスが不十分な場合。
覚えておくべきこと:
私どもは、FDAの510(k)の申請ルートを検討される際に、この記事が要求事項やその他の考慮事項を検討するうえで役立つことを願っています。
貴社の医療機器の実質的同等性の評価に関する詳しいガイダンスは、FDAのガイダンスを参照してください。